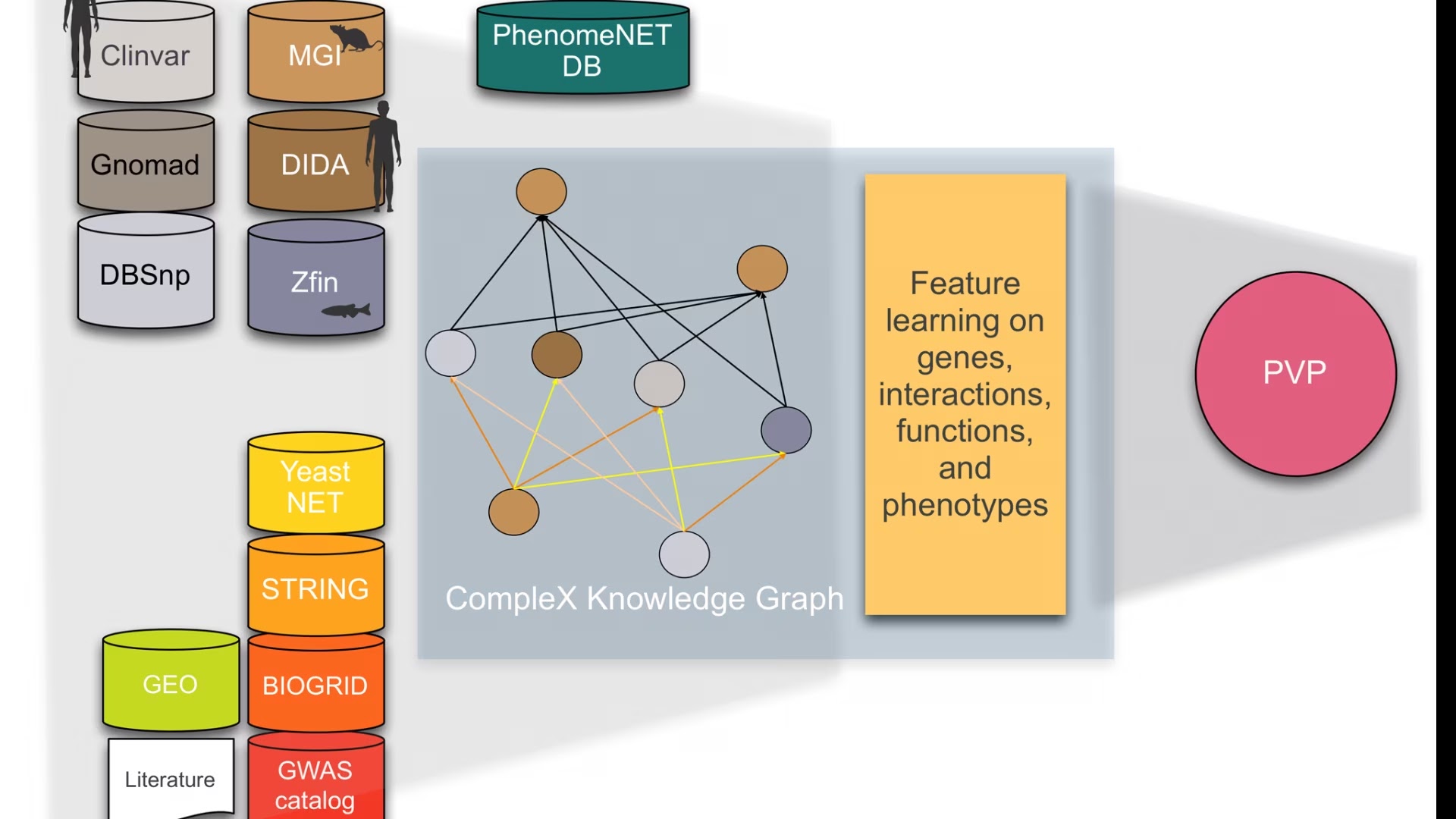
CompleX: Variant prioritization in complex disease
Phenotype-based methods have repeatedly shown to be highly effective in identifying causative variants in whole genome or whole exome sequences. The main limitation of phenotype-based methods, however, is the limited availability of characterised genotype-phenotype associations. Model organism phenotypes have in the past been used to supplement genotype–phenotype associations observed in humans and were demonstrated to predict disease genes. Nevertheless, in almost all cases, genotypes are loss-of-function or gain-of-function variants in single genes. Consequently, phenotypes that arise specifically from abnormal functioning of two or more genes in the same individual are not commonly captured; in the cases in which complex genotypes and their associations with phenotypes are recorded (e.g., in the mouse and fish model organism databases), they are not integrated, not distinguished by the type of interaction between variants, and cannot systematically be queried.

Neuro-symbolic systems
Symbolic methods and statistical connectionist methods are two main approaches to artificial intelligence. While symbolic methods are very widely used to represent knowledge in biology and biomedicine in the form of ontologies, only few methods have been developed that can utilize the information contained in these ontologies for building machine learning models. We work on methods that combine deductive inference and statistical models to improve knowledge representation and data analysis in biology. We use both syntactic and model-theoretic approaches.

Pathogen informatics
Understanding the relationship between the pathophysiology of infectious disease, the biology of the causative agent and the development of therapeutic and diagnostic approaches is dependent on the synthesis of a wide range of types of information. Provision of a comprehensive and integrated disease phenotype knowledgebase has the potential to provide novel and orthogonal sources of information for the understanding of infectious agent pathogenesis, and support for research on disease mechanisms. We are developing PathoPhenoDB, a database containing pathogen-to-phenotype associations. PathoPhenoDB relies on manual curation of pathogen-disease relations, on ontology-based text mining as well as manual curation to associate phenotypes with infectious disease. Using Semantic Web technologies, PathoPhenoDB also links to knowledge about drug resistance mechanisms and drugs used in the treatment of infectious diseases. We further work on exploiting pathogen-to-phenotype association in predictive models to understand host-pathogen interactions and generating new candidate drugs.
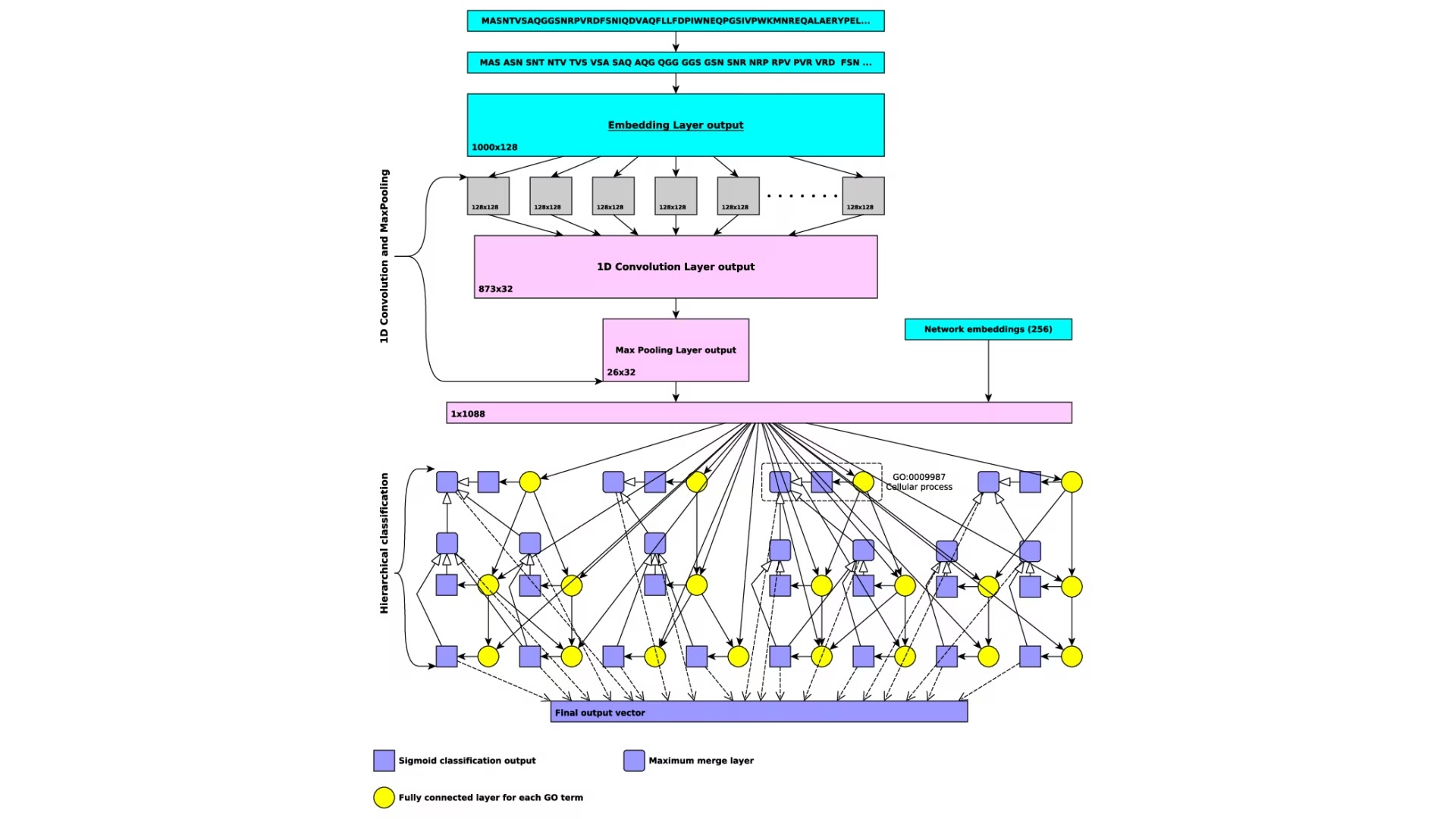
Prediction of functions and phenotypes
We will develop and expand novel methods for predicting protein functions and their loss of function phenotypes. We will utilize a deep neural network algorithm and combine them with symbolic inference into neural-symbolic algorithms. Our work will significantly extend our previously developed method for predicting protein functions called DeepGO through methodological advances in machine learning, incorporation of broader data types that may be predictive of functions, and improved systems for neural-symbolic integration.