Details
Infectious diseases are caused by a wide range of organisms (viruses, bacteria, fungi, worms, protozoa) that are generally considered as pathogens. Antimicrobial drugs are often the first-line therapy for infectious diseases. However, drug resistance accumulates over time due to the selection of genetic changes in pathogen populations when they are exposed to antimicrobial drugs (such as antibiotics, antifungals, antivirals, antimalarials, and antihelmintics). It now becomes crucial to develop strategies that can identify a pathogen rapidly and determine successful treatment options based on functional information in the pathogen relevant to drug resistance mechanisms.
While functional information about pathogens and their interactions with hosts is increasingly becoming available on a molecular level through large-scale studies, phenotypes observed in a patient are not only mediated through direct molecular interactions between a pathogen and host but also through the immune response and physiological and patho-physiological processes affecting the entire host organism. Phenotypes observed in a patient provide a readout for all these processes and generally provide a proxy for the mechanism through with pathogens elicit their signs and symptoms. While there is a wide range of phenotypes that are shared across multiple infectious diseases as a result of common immune system processes and immune response to pathogens, certain host-pathogen interactions may result in specific phenotypes through which pathogens can be broadly distinguished.
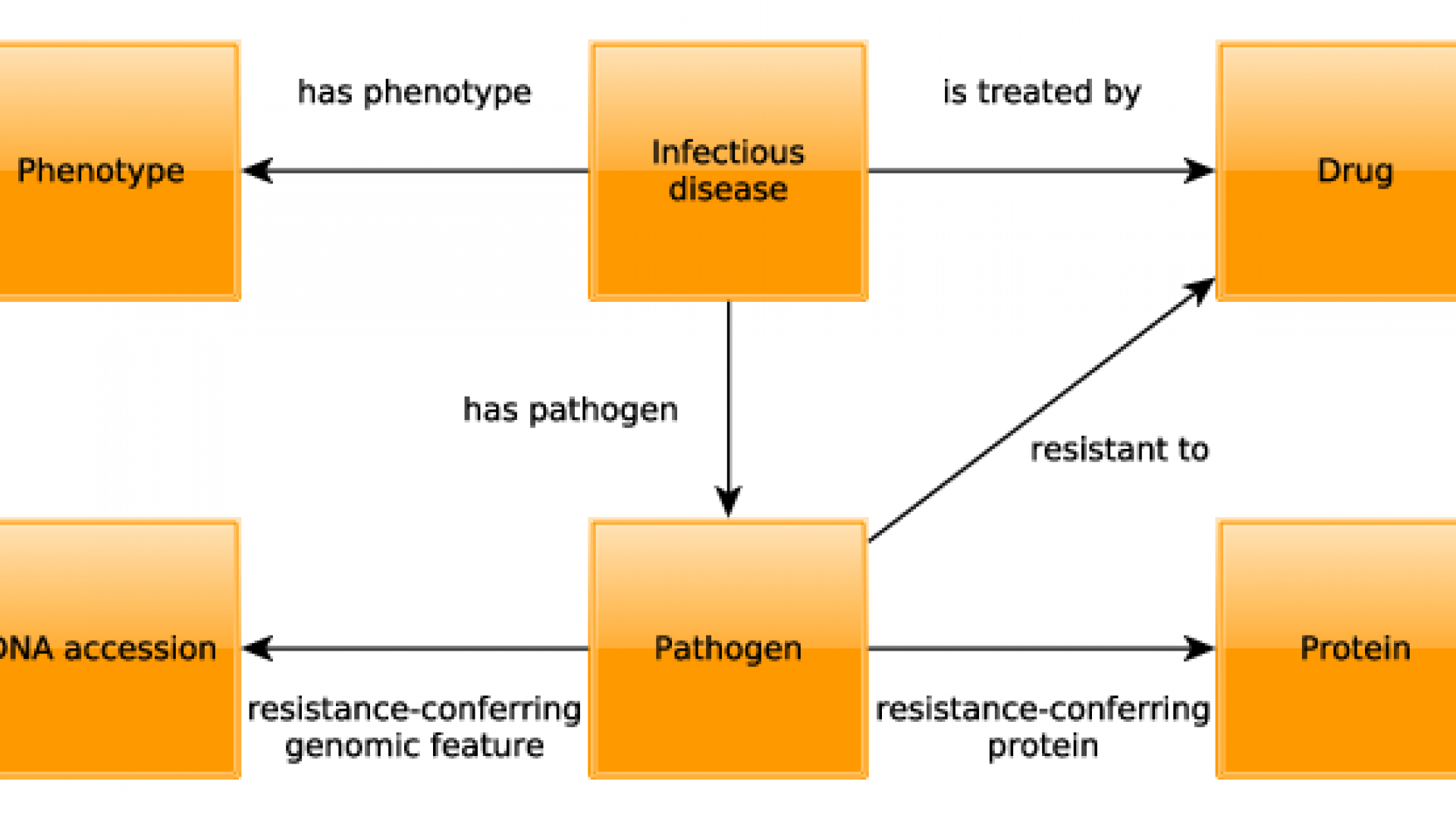
Phenotype-based computational analysis methods can uncover molecular mechanisms in Mendelian diseases and have been applied to the discovery of disease mechanisms from animal models and to the investigation of drug mechanisms and drug repurposing. In the area of infectious disease, similar methods may be applicable, mainly to investigate mechanisms of virulence and pathogenicity. Application of phenotype-based methods requires matching phenotypes observed in a particular physiological or pathological state with the phenotypes known to be associated with pathogens, and the use of this information to reveal molecular mechanisms. Currently, there is no comprehensive database of pathogen-to-phenotype associations that can be used for this purpose.
We have developed PathoPhenoDB, a database of pathogen-to-phenotype associations intended to support infectious disease research. PathoPhenoDB is a database that relies on pathogen–disease associations curated manually from public resources and the scientific literature. We further expanded the pathogen–disease associations by complementary text-mined data. PathoPhenoDB links pathogens to their phenotypes based on manually-curated and text-mined disease–phenotype associations. Furthermore, PathoPhenoDB links pathogens to drugs that are known to treat infections by the pathogen, and further links pathogens to drug resistance genes and proteins as well as to the drugs against which these genes or proteins convey resistance so that the information in PathoPhenoDB can be utilized directly for research on drug resistance mechanisms. PathoPhenoDB is freely available on http://patho.phenomebrowser.net, and the data can be obtained through a public SPARQL endpoint.
Identification of host-pathogen interactions (HPIs) can reveal mechanistic insights of infectious diseases for potential treatments and drug discoveries. Current computational methods for the prediction of HPIs often rely on our knowledge on the sequences and functions of pathogen proteins, which is limited for many species, especially for species of emerging pathogens. Matching the phenotypes elicited by pathogens with phenotypes associated with host proteins might improve the prediction of HPIs. We developed an ontology-based method that prioritizes potential interaction protein partners for pathogens using machine learning models. Our method exploits the underlying disease mechanisms by associating phenotypic and functional features of pathogens and human proteins, corroborated by multiple ontologies as background knowledge. Additionally,
by embedding the phenotypic information of the pathogens within a formally represented taxonomy, we demonstrate that our model can also accurately predict interaction partners for pathogens without known phenotypes, using a combination of their taxonomic relationships with other pathogens and information from ontologies as background knowledge. Our results show that the integration of phenotypic, functional and taxonomic knowledge not only improves the prediction of HPIs but also enables us to investigate novel pathogens in emerging infectious diseases.
Collaborators
- Paul Schofield, University of Cambridge