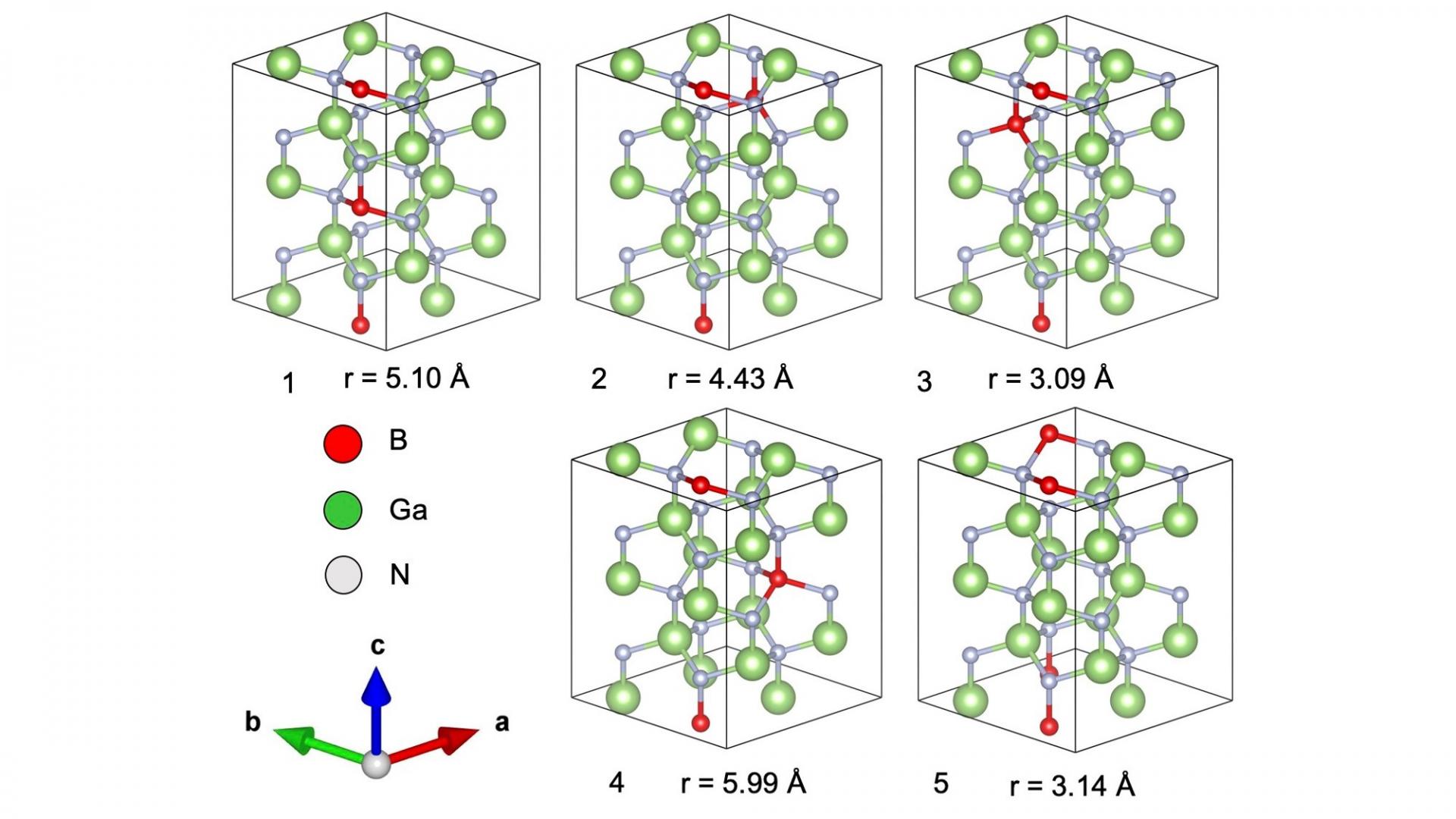
The paper reports an atomistic origin of composition pulling effect in all GaN-based ternary alloy was clarified from the the geometric configurations using first-principles calculations. We found that the most stable configuration involves two B, In, or Al atoms occupying Ga sites in wurtzite GaN along the c-axis which is the typical epitaxial growth direction.
[Abstract] Some fluctuations in composition are commonly observed in epitaxial-grown III-V multinary alloys. These fluctuations are attributed to compositional pulling effects and an insight on their atomistic origin is necessary to improve current epitaxial growth techniques. In addition, the crystallinity of III-V multinary alloys varies widely depending on the constituent atoms. Using first principles calculations, we then investigated different geometric configurations of gallium nitride (GaN)-based ternary alloy, X0.125Ga0.875N where X is the minority atom which is either boron (B), aluminum (Al), or indium (In). The minority atoms are presented as two atoms in the simulation cell, and the energetics of five geometric configurations are analyzed to estimate the most stable configuration. For the B0.125Ga0.875N alloy, the most stable configuration is the one where the minority atoms occupy gallium (Ga) sites in a collinear orientation along the c-axis. On the contrary, the configurations along the in-plane direction result in a higher energy state. In0.125Ga0.875N and Al0.125Ga0.875N also show the same trend with a small relative energy difference. These preferential sites of minority atoms are consistent with composition pulling effects in wurtzite nitride phases. Moreover, the degree of crystallinity for wurtzite nitride alloys can be well described by the order of calculated relative energy.
